
Update on Cancer Immunotherapy and Immuno Oncology
The past several years have been a particularly optimistic period for immuno-oncology. The first approval of modern cancer immunotherapy was interferon-alpha in 1986 for hairy cell leukemia, and later for chronic myelogenous leukemia, follicular non-Hodgkin lymphoma, melanoma, and AIDS-related Kaposi’s sarcoma (1) Several other agents have been approved since then, but a transformation in the landscape of Immuno Oncology (IO) started with the approval of ipilimumab—a checkpoint inhibitor targeting cytotoxic T-lymphocyte antigen 4 (CTLA- 4) for advanced melanoma in 2011(2)
Since then there has been an explosion of agents that work by enhancing body’s immune response to cancer. As of this writing there has been 26 immunotherapies approved in various cancers, and 17 types of cancer have at least one approved immunotherapy as a treatment option.

Approved Immuno Oncology agents against cancer. (Courtesy: Annals of Oncology)
In the past 3 years alone, five new checkpoint inhibitors (targeting PD-1 or PD-L1), two new cell therapies (targeting CD19), and one new CD3-targeted bispecific antibody (also targeting CD19) have been approved (3-6). Below is a brief summary of the clinical and scientific update on these exciting new class of molecules which has shown remarkable activity across a wide array of cancer types.
Checkpoint inhibitors
Immune cells navigate the body looking for anything that does not belong—bacteria, viruses, and even cancer cells. They do so by using their molecular receptors to scan for foreign molecules that intruder cells display on their surface. Once an intruder is detected, a class of immune cells, known as cytotoxic T cells, move in to eliminate it. Unfortunately, cancers have a number of ways to hide from immune cells and avoid their attack. One of the most successful classes of Immuno Oncology uses drugs that are known as immune checkpoint inhibitors.
“Check points” are certain proteins that down regulate the immune system, acting as a “break” and examples include the protein PD-1 (programmed cell death protein 1), PD-L1 (programmed death-ligand 1), PD-L2 (programmed death-ligand 2), and CTLA-4 (cytotoxic T-lymphocyte antigen 4 (CTLA-4). Why do our body need these “check points” that down regulate our immune system? Because body has to work hard to suppress its own immune reactions most of the time. The immune system has enough arsenal that it can kill us faster than whatever ails us, and in healthy individuals, these immune checkpoints prevent autoimmunity.
Many Cancers have unfortunately learned to exploit this check point pathways so it will be spared from destruction by the immune cells. The immune check point inhibitors are monoclonal antibodies against these “check points”, which work by taking “the brakes off” the immune system. Checkpoint-blockade immunotherapy has hence been the most exciting advance made in cancer treatment in recent years and has now become a new arsenal in the fight against several cancers, often showing dramatic response even in advanced stages.
The science behind Immune Oncology:
It has been known for decades that immune surveillance is critical against the development of various malignancies and patients with impaired immune system are at high risk for the development of cancer. It has also been known for more than 50 years that the human immune system has the potential to be a very powerful anticancer therapy. Part of the rationale for doing bone marrow transplantations was to give the patient a new immune system, which could help fight the patient’s cancer, with the so called graft versus leukemia / lymphoma effect, and that’s where so much interest in cellular immunotherapy originated.
In many cancers, oncogenesis is accompanied by the accumulation of mutations, which can provide a selective advantage to populations of cancer cells by increasing their degree of genetic diversity and accelerating their evolutionary fitness. Yet this diversity comes at a cost to the cancer cell: the further a cancer cell diverges from a normal cell, the more likely it is to be recognized as foreign by the immune system. Although long considered a possibility, it has been demonstrated only in the past five years that the mutational burden of tumors contributes to immune recognition of cancer and that it may, at least partly, determine a person’s response to cancer immunotherapy (7)
The role of the immune system in cancer remained unappreciated for many decades because tumors effectively suppress immune responses by activating negative regulatory pathways (above described checkpoints) that are associated with immune homeostasis or by adopting features that enable them to actively escape detection (8) Two such checkpoints, cytotoxic T-lymphocyte protein 4 (CTLA4) and programmed cell death protein 1 (PD-1), are the most studied so far.
CTLA4 is a negative regulator of T cells, essentially acting as a break on T-cell activation. The cell-surface receptor PD-1 is expressed by T cells on activation during priming or expansion and binds to one of two ligands, PD-L1 and PD-L2. Many types of cells can express PD-L1, including tumor cells and immune cells after exposure to cytokines such as interferon (IFN)-γ; however, PD-L2 is expressed mainly on dendritic cells in normal tissues. Binding of PD-L1 or PD-L2 to PD-1 generates an inhibitory signal that attenuates the activity of T cells.
Both CTLA4 and the PD-1/PD-L system thus acts as “immune check points”, or “breaks” in the host immune response. Although it was found that blocking these check points can elicit antitumor response in mice as early as 1996 (9) it has been only in the last couple of years its role and significance found in humans. But once this flood gate has been opened, the development has been swift, and more marked than the development of any dug class against cancer. And response have been observed across a wide variety of cancer types, unlike traditional targeted therapies where the effect is often limited to a small subset of patients. (10)
Even more importantly, the responses are often durable, lasting years or indefinitely, and occur without causing serious toxicity in most people. These results suggest that many people with cancer have pre-existing T-cell mediated immunity that is restrained by the PD-L1/PD-1-induced suppression of T cells. They also emphasize the role of immunosuppression as a main impediment to the series of steps that is required for effective anticancer responses — the cancer–immunity cycle (11)
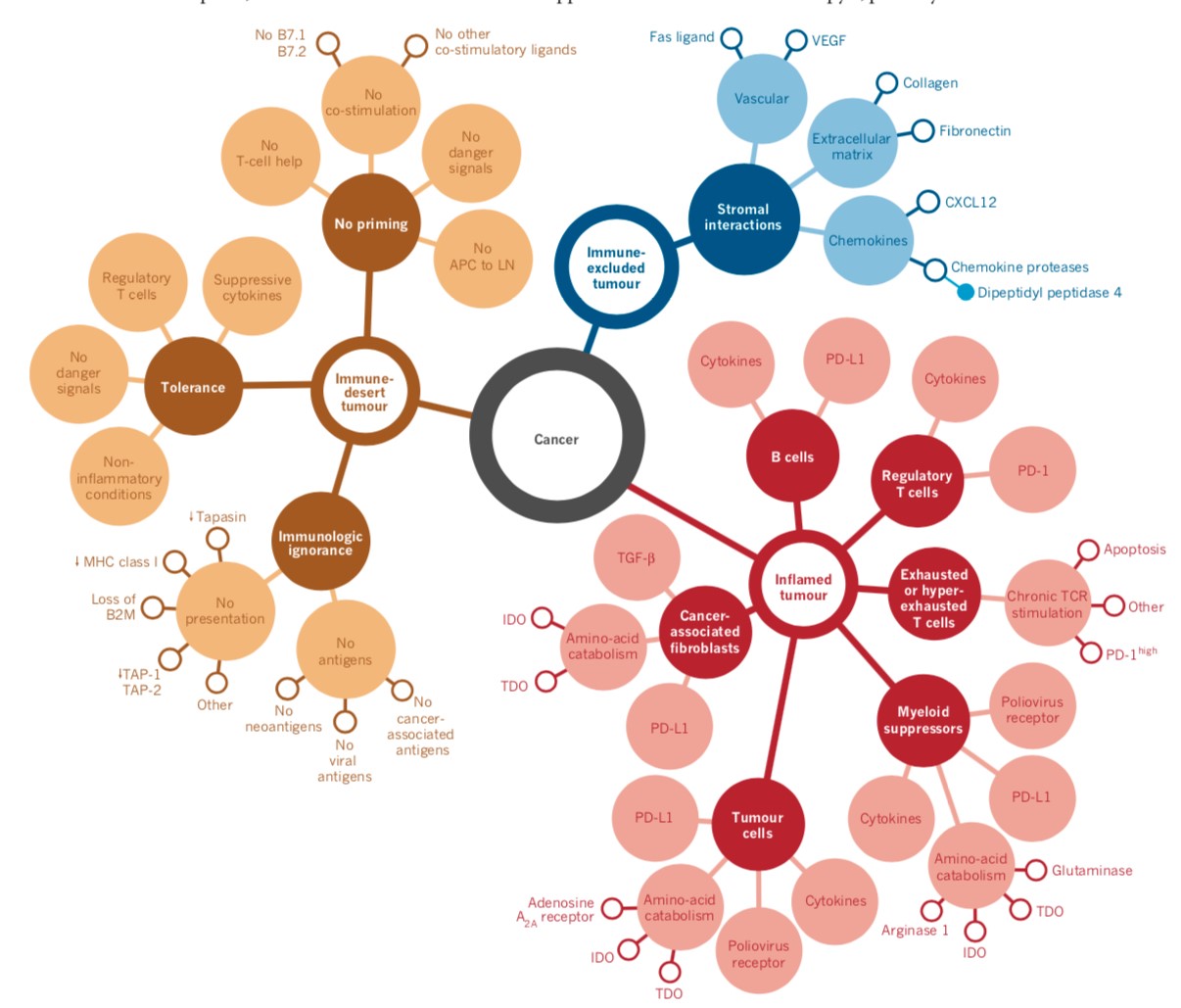
Cancer Immune Phenotype (Image Courtesy: Nature)
Anticancer immunity in humans can be segregated into three main phenotypes: the immune-desert phenotype (brown), the immune–excluded phenotype (blue) and the inflamed phenotype (red). Each is associated with specific underlying biological mechanisms that may prevent the host’s immune response from eradicating the cancer. A tumor that is characterized as an immune desert can be the result of immunological ignorance, the induction of tolerance or a lack of appropriate T-cell priming or activation. Immune-excluded tumors may reflect a specific chemokine state, the presence of particular vascular factors or barriers, or specific stromal-based inhibition.
Inflamed tumors can demonstrate infiltration by a number of subtypes of immune cells, including immune-inhibitory regulatory T cells, myeloid-derived suppressor cells, suppressor B cells and cancer-associated fibroblasts. Tumor-infiltrating lymphocytes that express CD8 may also demonstrate a dysfunctional state such as hyper exhaustion. Tumor cells in inflamed tumors can also express inhibitory factors, down regulating MHC class I molecule expression or other pathways that de-sensitize them to anticancer immunity.
The profile that responds best to immunotherapy are the so called immune-inflamed phenotype (Figure 2 above), which is characterized by the presence in the tumor parenchyma of both CD4- and CD8-expressing T cells, often accompanied by myeloid cells and monocytic cells; the immune cells are positioned in proximity to the tumor cells. (12) This profile suggests the presence of a pre-existing antitumor immune response that was arrested — probably by immunosuppression in the tumor bed.
The second profile is the immune-excluded phenotype, which is also characterized by the presence of abundant immune cells. However, the immune cells do not penetrate the parenchyma of these tumors but instead are retained in the stroma that surrounds nests of tumor cells. The stroma may be limited to the tumor capsule or might penetrate the tumor itself, making it seem that the immune cells are actually inside the tumor. After treatment with anti-PD-L1/ PD-1 agents, stroma-associated T cells can show evidence of activation and proliferation but not infiltration, and clinical responses are uncommon. These features suggest that a pre-existing antitumor response might have been present but was rendered ineffective by a block in tumor penetration through the stroma or by the retention of immune cells in the stroma. T-cell migration through the tumor stroma is therefore the rate-limiting step in the cancer–immunity cycle for this phenotype. (13)
The third profile, the immune-desert phenotype, is characterized by a paucity of T cells in either the parenchyma or the stroma of the tumor. Although myeloid cells may be present, the general feature of this profile is the presence of a non-inflamed tumor microenvironment with few or no CD8-carrying T cells. Unsurprisingly, such tumors rarely respond to anti-PD-L1/PD-1 therapy. This phenotype probably reflects the absence of pre-existing antitumor immunity, which suggests that the generation of tumor-specific T cells is the rate limiting step. The immune-desert phenotype and the immune-excluded phenotype can both be considered as non-inflamed tumors.
Predicting response to immunotherapy:
The immune-inflamed phenotype correlates generally with higher response rates to anti-PD-L1/PD-1 therapy, which suggests that biomarkers could be used as predictive tools. Most attention has been paid to PD-L1, which is thought to reflect the activity of effector T cells because it can be adaptively expressed by most cell types following exposure to IFN-γ6, (14). In an increasingly large clinical data set, it is becoming clear that the expression of PD-L1 in pretreatment biopsies facilitates enrichment with people who are most likely to respond to antibodies against PD-L1 or PD-1. PD-L1 expression also correlates strongly with various markers of active cellular immunity, including IFN-γ, granzymes, CXCL9 and CXCL10. The presence of these biomarkers or others such as T cells that carry the CD3 antigen or tumor mutational burden may also enrich for responders. When used in combination with PD-L1 expression, these biomarkers may enhance predictive power (15)
As described previously, it is probable that the mutation burden of a given tumor will contribute to its immune profile with clearest association demonstrated between response and overall mutational burden. The greater the number of mutations in a given tumor, the more probable it is that some of the mutations will be immunogenic, providing targets for T-cell attack (16). Mutations that arise early in oncogenesis and are shared by almost all of the cancer cells in an individual (known as truncal mutations) may generate more effective anticancer T-cell responses than mutations that arise later on and are limited to only a subpopulation of cancer cells (known as branch mutations)
The importance of the cancer–immune set point
The cancer–immune set point is the threshold that must be overcome to generate effective cancer immunity. The set point can be understood as a balance between the stimulatory factors (Fstim) minus the inhibitory factors (Finhib), which together must be equal to or greater than 1, over the summation of all T-cell antigen receptor (TCR) signals for tumor antigens. The cancer–immune set point is shown as:
∫ (Fstim) − ∫ (Finhib) ≥ 1 ∕ ∑ n=1, y (TCRaffinity × frequency) (17)
It is probable that –immune set point of a particular person is already determined by the time of clinical presentation, driven by the inherent immunogenicity of the tumor and by the responsiveness of the individual’s immune system. Although it is reasonable to assume that various lines of cancer therapy or changes in environmental factors might alter Fstim and Finhib, such changes might only be transient.
Often, the set point that is identified using pretreatment biopsies is similar to the set point determined by biomarker profiling from biopsies taken on progression after therapy. Likewise, despite the continued accumulation of mutations in a tumor as a function of time, primary and metastatic lesions can exhibit similar immune profiles. The features that determine the set point may therefore reflect genetic factors that are specific to a given tumor, the genetics of the person with cancer, or the extent to which antitumor immunity had developed initially.
Although largely conceptual, the idea of a set point provides a framework to help organize the torrent of clinical and biomarker data that will emerge over the coming months and years. The number of targets that could prove effective for cancer immunotherapy is great; the number of potential combinations of therapeutic agents that are directed against these targets (or combinations of such agents with conventional standard-of-care agents) is even greater. The development of some cancer therapies may be largely empirical, but it can be guided by considering, even in general terms, the elements that comprise cancer immunity.
Adoptive cellular Immunotherapy:
Apart from immune check point inhibitors, another area of rapid development in immune oncology has been the use of CAR-T cells. Adoptive cell immunotherapy boosts the body’s immune defenses against cancer in a completely different way—by genetically re-engineering a patient’s own immune T cells. It was Dr. Carl June and team from University of Pennsylvania who was the first to experiment with genetically reprogramming T cells, now known as CAR T cells. (18)
CAR T cells are custom made to work against the cancer in each individual patient. To create these cells, researchers collect immune T cells from the patient and insert an artificial gene into the cells. The gene is designed to endow T cells with chimeric antigen receptors that can detect unique molecules on cancer cells after CAR T cells are multiplied in the laboratory and injected back into the patient. In essence, CAR T-cell therapy is both a gene therapy and an immunotherapy.
When the CAR T-cell receptor attaches to a molecule on a cancer cell, it sends a signal to turn on the destruction machinery of the T cell. Unlike traditional cancer treatments, this living therapy needs to be given to the patient only once, because CAR T cells continue to multiply in the patient’s body. As a result, the anticancer effects of CAR T cells can persist and even increase over time.
Challenges
Immuno Oncology brings its own unique set of challenges. Selecting the appropriate patients for the highly expensive therapies are still not completely well defined. While markers like PD-L1 expression can act as a good predictor of response, the predictive value varies between various tumor types. While the response to these drugs could be dramatic some time even in terminally ill patients, the exorbitant cost prevents its use more widely in deserving patients. Not all patients respond to these medications, and in a sub group of patients the cancer cells could become resistant after a period of initial response. The mechanisms of resistance remains an active area of intense research but as of this writing clinical options for patients who progress on immunotherapy remains limited. (19)
Immunotherapy drugs also come with a unique set of side effects, and clinicians should familiarize with these often unusual side effects so that it could be recognized early, as some of the side effects could be serious, even fatal (20)
Another challenge for future drug development is the identification of the appropriate targets on cancer cells. The problem with many cancers is that they are too close to self and there aren’t easily identifiable target antigens that allow you to apply immune therapy without off-target toxicity. It is quite possible that over the next 5 to 10 years, some of the biggest advances will come from identifying new targets for various kinds of cancers and then applying vaccine therapy, antibody therapy, and cellular therapy against these specific targets. This will be done through extensive studies of cancer cell biology, DNA sequencing, and other techniques that will allow this type of immunotherapy to become increasingly personalized to a patient’s specific tumor and the tumor’s specific targets.
It’s going to be a long time before chemotherapy is obsolete. Some chemotherapy may actually have a role concurrently with immunotherapy and may function to modulate the immune interactions. There will also be instances where we use multimodality therapy, including chemotherapy, radiation therapy, and immunotherapy.
Curing Cancer
Even the most successful targeted therapies like imatinib for chronic myeloid leukemia are rarely curative, and once the therapy is withdrawn the cancer could come back. Immunotherapy however could be curative even in some advanced malignancies. Going back to the example of bone marrow transplantation, there are circumstances when patients with leukemia or lymphoma relapse after allogeneic transplant and can be cured with infusions of normal T cells from their original transplant donor (donor lymphocyte infusion). There has been cases of patients with advanced malignancies who had received check point inhibitors or CAR-T cell therapy who have been cured and remains disease free despite not receiving any ongoing therapy.
Immune therapies are the new frontier in cancer therapy, and the field is in its infancy. As of this writing, there were 940 IO agents in clinical development, with another 1064 in preclinical phase, with over 3000 interventional active clinical trials evaluating these clinical-stage immunotherapies with a target of enrolling over half a million patients across the world. So it can be anticipated that the application of immunotherapy in the treatment of cancer will grow dramatically over the next decade.















